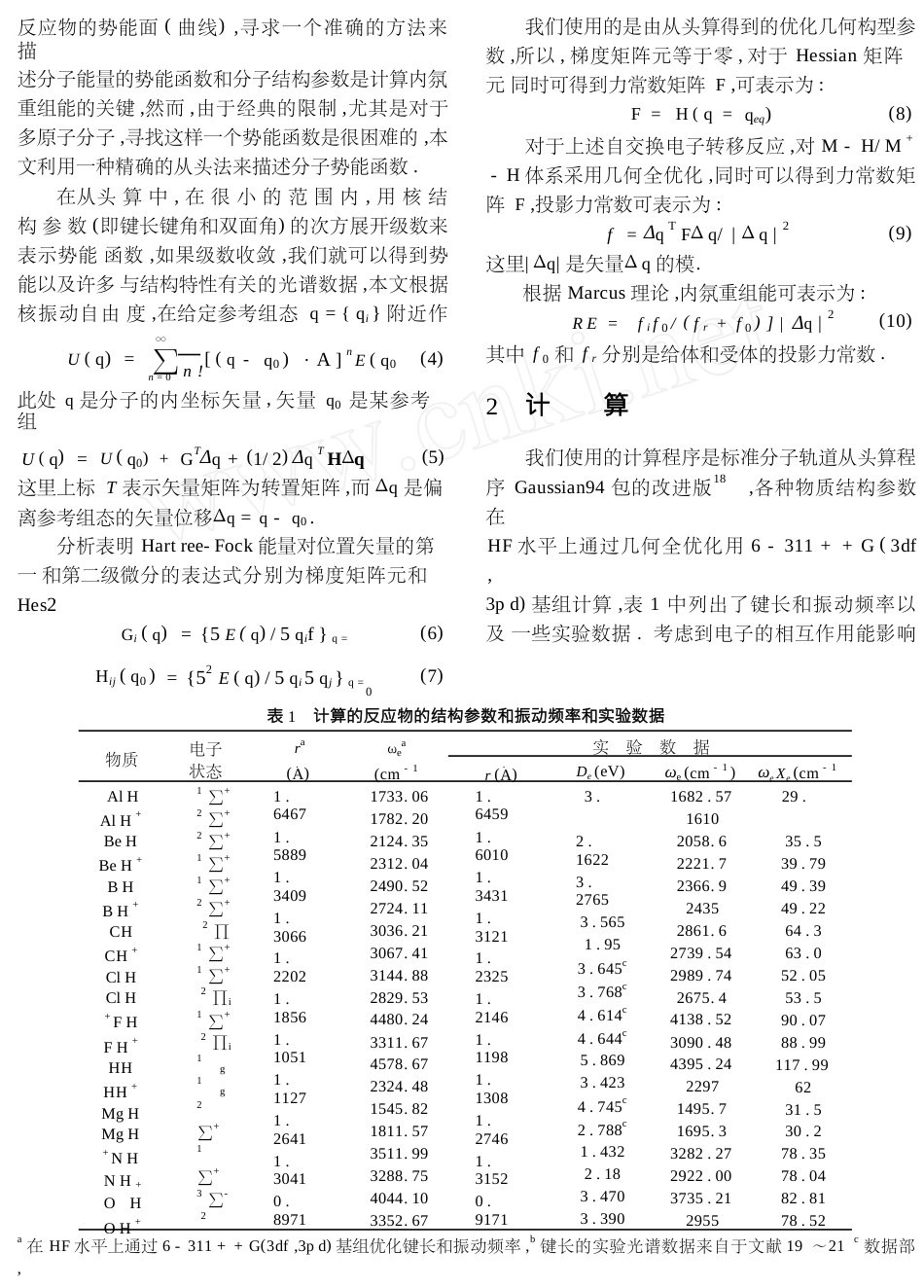
第26卷第1期2000年1月曲阜师范大学学报JournalofQufuNormalUniversityVol.26No.1Jan.2000ΞM+M-H/-H体系重组能的从头算法段春生①,吴月顺①,李凤华②,张传松②,邢玉梅②,邢丽②,周正宇②(第一作者:男,40岁,讲师;①青岛大学师范学院化学系,266023,山东省青岛市;②曲阜师范大学化学系,273165,山东省曲阜市)摘要:基于一种新的从头算法计算了M-H/M+-H体系电子转移反应的重组能,对每个反应物进行几何优化.结果表明:用从头算法计算的双原子分子的键长和振动频率与实验光谱数据吻合较好.利用精确的重组能George-Griffith-Marcus(GGM)模型计算得到的重组能数值与实验光谱数据中得到的值相比较,结果显示,在气相状态的电子转移反应中,直接计算重组能的值要比传统的GGM方法准确.关键词:从头算法;重组能;电子转移反应中图分类号:O641.13文献标识码:A文章编号:1001-5337(2000)01-0064-04电子转移反应是一种重要的化学过程,并且渐渐成为理论和实验研究的焦点1~10.重组能是一个表征电子转移过程动力学的重要参数,是决定反应速度的重要因素.它表示一种能量变化,这种变化与反应物的结构变化以及反应物由始态到过渡态或终止态所必须的介质环境有关.通常人为地将重组能划分为内氛和外氛两种贡献.外氛贡献将宏观溶剂作为连GGM模型是重组能精确定义的简化结果.本文基于重组能的精确定义计算气态M-H/M+-H体系中电子转移反应的内氛重组能,运用从头法计算Hartree-Fock自洽场内氛重组能.将精确的势理论方法1根据电子从供体转移到受体先于分子结构重组的假设以及重组能的精确定义式,即重组能就是受体接受电子时的非平衡态到接受电子后到达新平衡态的能量降低值加上供体从失去电子时的非平衡态到失电子后到达新的平衡态的能量降低值,对于下列伴随零自由能变的热电子自交换反应:算得11,12.对于气相反应体系,由于反应物和周围介质的作用力非常弱,使外氛重组贡献几乎等于零,因此可以忽略.内氛结构重组的影响支配着整个电子转移过程.所以,对于气相电子转移反应,内氛重组能的精确求解就显得尤为重要.在20世纪50年代George-Griffith提出自交换模型14后,前人又相继提出了改进George-Griffith公式中势能常量因子特性的各种方法,例如,在George-Griffith工作的基础上,Marcus对内氛重组能提出了一个概括性的表M+-H+M-HΖM-H+M+-重组能REi可表示为:REi=ΔRE1+ΔRE2(1)+M-HM-HΔRE1-H(r)-H(r)(2)(3=EM+-EM+述12,13.在这个模型中,测定的内氛重组能,需要计+EM-H(rM-HM-H)EM-H(r)ΔRE2=-算键的力常数.后来,利用锁定偶极取向(LDO),改进的平均偶极取向(IADO),半经典的微扰转动状态(PRS)模型15,16,以及分子轨道(MO)微分重叠的中间忽略17测定键力常数和内氛重组能.但这些方法都基于GGM自交换模型,即这个模型仅仅适用于谐振子势能,而且活化物在整个电子转移过程中必eq其中ΔRE1是在电子从M-H转移到M+-H后立即产生M+-H的重组能,rM-H表示M-H的平eq+rM-H+均键长,而是M-H的平均键长.eq上面的方程式表示,当一个电子绝热地从给体转移到受体时,体系从非平衡态发生弛豫到平衡核组态,体系弛豫过程的能量变化就相当于重组能.很显然,内氛重组能的计算很大程度上依赖于Ξ收稿日期:1999—06—18基金项目:国家自然科学基金(29673025)山东省自然科学基金(反应物的势能面(曲线),寻求一个准确的方法来描述分子能量的势能函数和分子结构参数是计算内氛重组能的关键,然而,由于经典的限制,尤其是对于多原子分子,寻找这样一个势能函数是很困难的,本文利用一种精确的从头法来描述分子势能函数.在从头算中,在很小的范围内,用核结构参数(即键长键角和双面角)的次方展开级数来表示势能函数,如果级数收敛,我们就可以得到势能以及许多与结构特性有关的光谱数据,本文根据核振动自由度,在给定参考组态q={qi}附近作Taylor级数展我们使用的是由从头算得到的优化几何构型参数,所以,梯度矩阵元等于零,对于Hessian矩阵元同时可得到力常数矩阵F,可表示为:F=H(q=qeq)(8)对于上述自交换电子转移反应,对M-H/M+-H体系采用几何全优化,同时可以得到力常数矩阵F,投影力常数可表示为:f=ΔqTFΔq/|Δq|2(9)这里|Δq|是矢量Δq的模.根据Marcus理论,内氛重组能可表示为:RE=fif0/(fr...