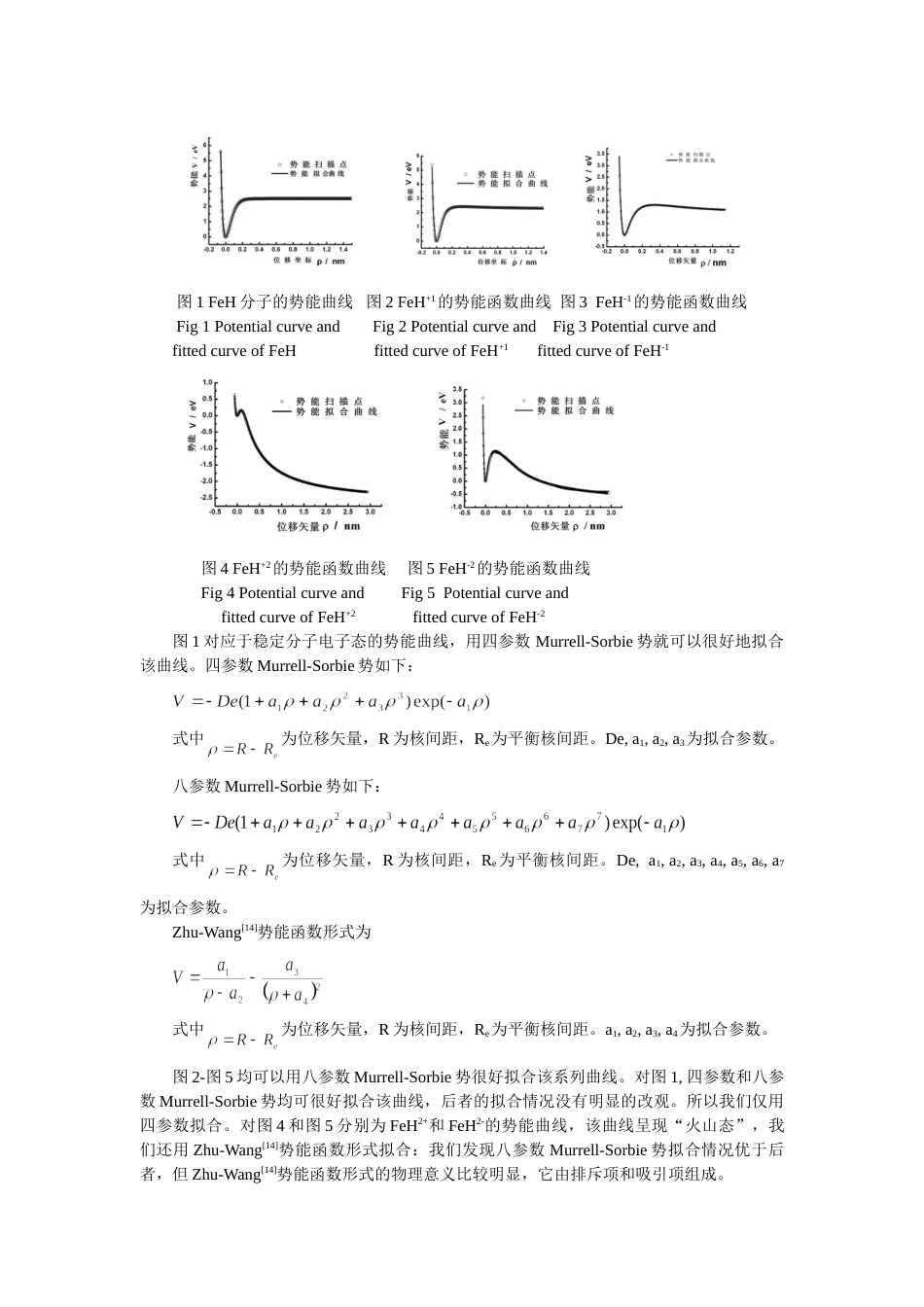
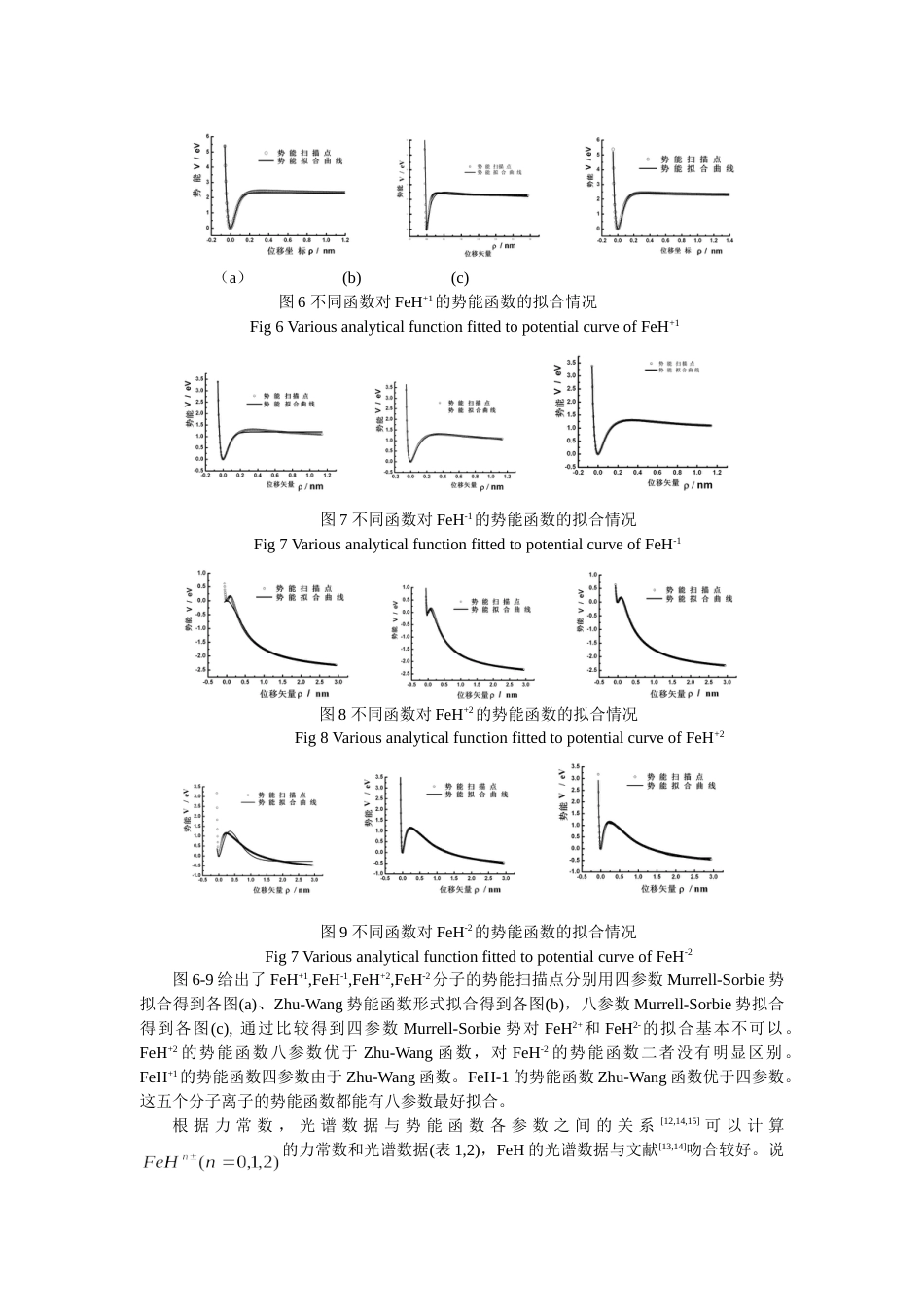
电荷对分子离子的势能函数和能级的影响谌晓洪张乐情王玲兰杰西华大学物理与化学学院成都610039用密度泛函B3lyp/6-311++g(d,p)方法对分子离子进行理论研究。结果表明:FeH,FeH+,FeH-1均能稳定存在,FeH+2和FeH-2有亚稳定态存在,其基态电子态分别是:4Δ(FeH),5Σ(FeH+),5Σ(FeH-),4Σ(FeH+2)和6Σ(FeH-2),FeH+2和FeH-2的势能函数呈明显的‘火山态’型,导出了相应的分子离子的解析势能函数、光谱数据和力常数,比较了四参数、八参数Murrell-Sorbie势和Zhu-Wang势对不稳定分子势能函数的拟合情况,指出了用八参数Murrell-Sorbie势对‘火山态’型势能函数的拟合也是合适的;同时讨论了电荷对势能函数和能级的影响。关键词:分子离子,密度泛函,势能函数,能级:O561.1;O561.4文献标识码:ATheeffectofchargeonthepotentialenergyfunctionandenergylevelforChenXiao-hongZhangLe-qingWangLingLan激eTheSchoolofPhysicsandChemistry,XihuaUniversity610039Chengdu,ChinaAbstract:Atheoreticalstudyonusingdensityfunctionalmethod(B3lyp/6-311++g(d,p))showthatFeH,FeH+,andFeH-arestable;FeH2±aremeta-stable.Theirelectronicstatesofgroundstateare4Δ(FeH),5Σ(FeH+),5Σ(FeH-),4Σ(FeH+2)and6Σ(FeH-2),Obviously,theenergycurvesofFeH2±havebothminimumandmaximum,whichareso-call“energytrapped”molecule.Murrell-SorbiefunctionswithFour-andEight-parametersandZhu-Wangpotentialhavebeenusedtofittheenergycurveswhichhavebothminimumandmaximum,theresultsshowthat,Eight-parametersMurrell-Sorbiefunctioncanfitthecurveswell.Atthesametime,theeffectofchargeonthepotentialenergyfuctionsandenergylevelsforarediscussed.Keywords:molecularions,densityfunctionaltheory,potentialenergyfunction,energylevel1引言由于FeH基团在第一周期过渡金属氢化物中有较复杂的电子结构,它是有相当意义的星际分子[1],铁还可以做为储氢材料[2]。所以对FeH分子的势能函数和光谱数据的正确理解,是有必要的。由于FeH的复杂的电子结构,实验和理论研究都有一定的困难。从实验上讲,有必要观察和解释它的光谱,进一步从理论上了解其内部电子运动。在宇宙中,氢和铁都有较高的丰度,FeH分子有可能在星际大气中存在[3-5]。二十世纪五十年代,在实验室根据观察到的近红外989.6nm的谱线,首先确认了FeH分子的存在[6]。1972年CarrollandMcCormack[5]观察到绿和蓝色区的两个光谱带,被认为是属于FeH分子的。1973年KlynningandLindgren[7]观察了高精度的近红外989.6nm谱线,1976年Carrolletal.[4,8]发表了FeH和FeD的电子谱。1979年Dendramis用基质隔离技术研究了FeH和FeD的振动光谱。大量的实验研究FeH分子外,理论研究也有不少。1988年Bauschlicher和Langhoff[10]首先用从头计算确定了该分子的基态。Langhoff和Bauschlicher[11]用CASSCF/MCRI方法全电子基组研究了该分子的基态和激发态。但是关于FeHn±(n=0,1,2)分子离子的势能函数的研究未见报道。本文用gaussian03软件包的B3lyp方法,在6-311++g(d,p)基组水平上研究了该系列分子离子基态和少数低激发态的结构,分析势能函数,能级以及电荷对势能函数和能级的影响。2分子离子的离解极限分子势能函数对应一定的电子状态,为了准确地表达体系的势能函数,需要首先确定正确的离解极限和可能的电子状态,Fe原子的基态电子态为5Dg,H原子的基态电子态为2Sg,根据原子分子反应静力学原理[12],由分离原子法可以构造出FeH分子的可能状态。异核双原子分子属于C∞v群,将原子群的表示分解为C∞v群的表示,直积和约化可得到C∞v群的不可约表示,从而得到FeH分子的可能电子状态。5Dg和2Sg分解为C∞v群的表示为,四重态,六重态均为FeH分子可能的电子状态,用B3lyp/6-311++g(d,p)对FeH分子可能电子态进行优化,结果表明FeH分子的基态为:4Δ,但6Σ的能量比前者仅高0.5eV,理论上讲它们都可能在实验观察中出现。而FeH分子可能的电子状态中包含了4Δ态,因此,FeH(X4Δ)可按分离原子法指出的方式离解,而2Sg为H原子的基态,5Dg为Fe原子的基态...