
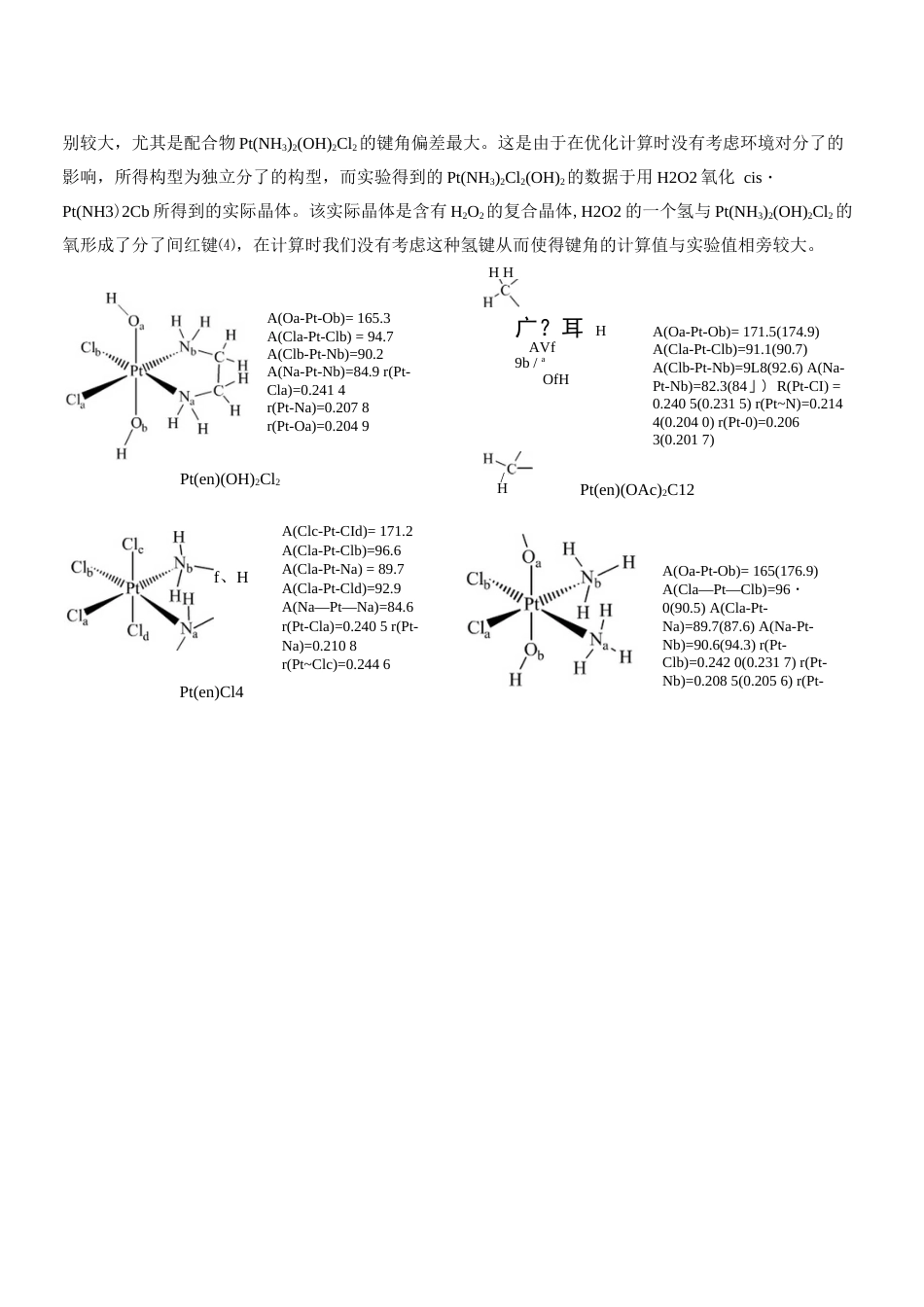
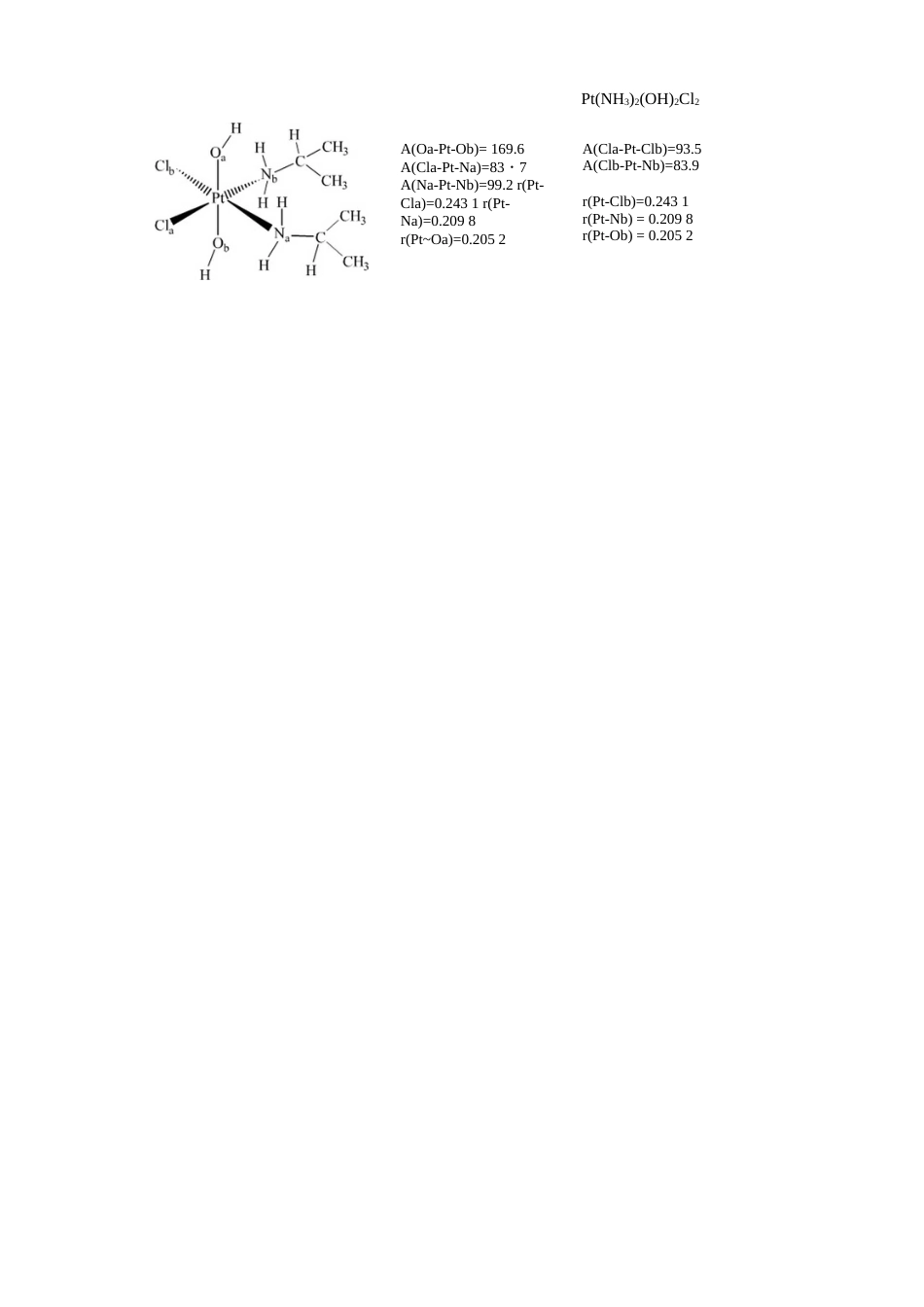

1、当您付费下载文档后,您只拥有了使用权限,并不意味着购买了版权,文档只能用于自身使用,不得用于其他商业用途(如 [转卖]进行直接盈利或[编辑后售卖]进行间接盈利)。
2、本站不对文档的完整性、权威性及其观点立场正确性做任何保证或承诺!文档内容仅供参考,付费前请自行鉴别。
3、如文档内容存在侵犯商业秘密、侵犯著作权等,请点击“举报”。
1、问:已经付过费的文档可以多次下载吗?
答:可以。登陆您已经付过费的账号,付过费的文档可以免费进行多次下载。
2、问:已经付过费的文档不知下载到什么地方去了?
答:电脑端-浏览器下载列表里可以找到;手机端-文件管理或下载里可以找到。
如以上两种方式都没有找到,请提供您的交易单号或截图及接收文档的邮箱等有效信息,发送到客服邮箱,客服经核实后,会将您已经付过费的文档即时发到您邮箱。
注:微信交易号是以“420000”开头的28位数字;
支付宝交易号是以“2024XXXX”交易日期开头的28位数字。
客服邮箱:
所有的文档都被视为“模板”,用于写作参考,下载前须认真查看,确认无误后再购买;
文档大部份都是可以预览的,笔杆子文库无法对文档的真实性、完整性、准确性以及专业性等问题提供审核和保证,请慎重购买;
文档的总页数、文档格式和文档大小以系统显示为准(内容中显示的页数不一定正确),网站客服只以系统显示的页数、文件格式、文档大小作为依据;
如果您还有什么不清楚的或需要我们协助,可以联系客服邮箱:
常见问题具体如下:
1、问:已经付过费的文档可以多次下载吗?
答:可以。登陆您已经付过费的账号,付过费的文档可以免费进行多次下载。
2、问:已经付过费的文档不知下载到什么地方去了?
答:电脑端-浏览器下载列表里可以找到;手机端-文件管理或下载里可以找到。
如以上两种方式都没有找到,请提供您的交易单号或截图及接收文档的邮箱等有效信息,发送到客服邮箱,客服经核实后,会将您已经付过费的文档即时发到您邮箱。
注:微信交易号是以“420000”开头的28位数字;
支付宝交易号是以“2024XXXX”交易日期开头的28位数字。





